

Density Functional Theory
Electronic, kinetic and thermodynamic properties: Dielectric breakdown at metal-oxide interfaces
- We study the electronic structure and local pressure of Al/crystal-SiO2 (Al/c-SiO2) and Al/amorphous-SiO2 (Al/a-SiO2) interface systems in the presence of oxygen vacancy.
- We found that oxygen vacancies close to the interface do not change the band offset or electronic structures. However, oxygen vacancies far away from the interface generate shallow hole trapping states.
- We also applied the Quantum Stress Density theory to calculate the local hydraulic pressure around each host oxygen atom to be removed for creating an oxygen vacancy.
- Our data also suggest strong correlations among local pressure, vacancy formation energy, electronic potential barrier height, and electronic hopping integrals, providing a unique yet comprehensive understanding of electronic properties on the metal/oxide interface.
- Our research represents an important milestone in the ultimate goal of an advanced understanding of dielectric breakdown
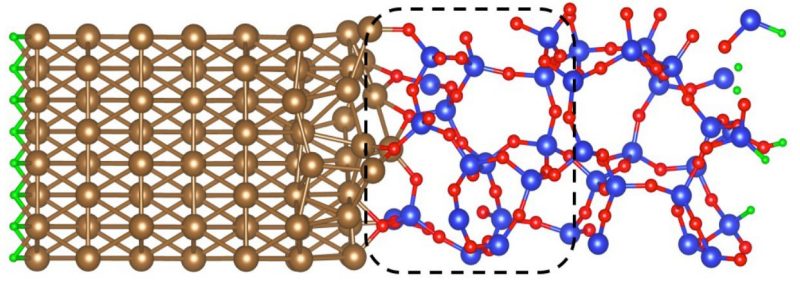
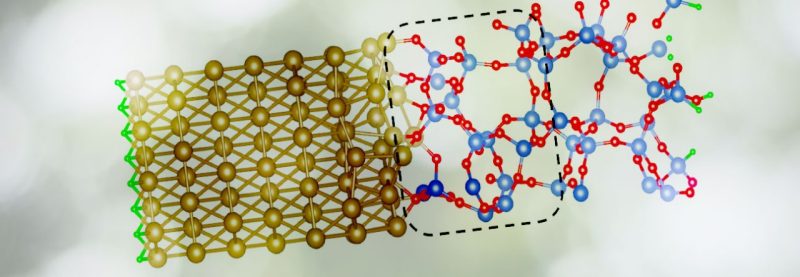
The picture shows Al/amorphous-SiO2 interface. The blue, red, gold, and green balls represent Si, O, Al, and H atoms, respectively. The dashed-line boxes indicate regions where oxygen vacancies have been created.
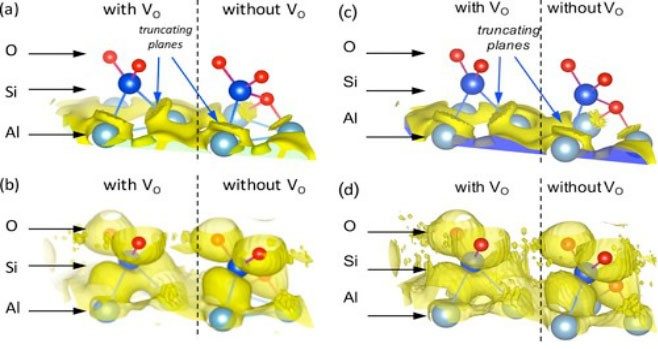
((a) and (c)) Charge density iso-surfaces in the interfacial Al Bader volumes for LDA and PBE0, respectively. ((b) and (d)) Electron localization function at the interface displayed at η= 0.5
for LDA and PBE0, respectively.
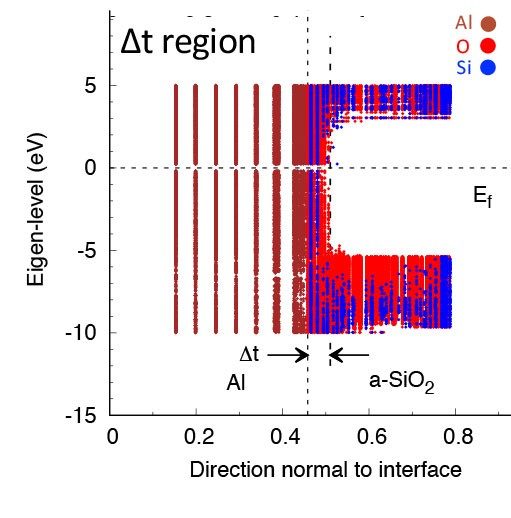
Spatially dependent band diagram of Al/silica amorphous systems in presence of Vo at the (∆t) region.
Electronic, kinetic and thermodynamic properties: thermal conductivity of metallic fuels
- Metallic Uranium (U) alloys are promising materials for use as nuclear fuels in thermal reactors and for Generation IV fast nuclear reactor designs.
- Understanding and properly modeling the thermal properties of U metal is a prerequisite for the study of U based metallic fuel alloys. In this work, we report a methodology to calculate the thermal conductivity of U metal using density functional theory (DFT).
- A Plane Augmented Wave (PAW) dataset within the local density approximation (LDA) was developed, the electron phonon scattering time was evaluated using the dataset, and finally the electron thermal conductivity was calculated by solving the Boltzmann transport equation.
- The calculated values taking into account only the electron-phonon interactions have good qualitative agreement with available experimental data.
- One of the major advantages of this work results in the absence of fitting parameters, so the approach can be readily extended to model thermal conductivity of metallic uranium alloys, both with and without point defects such as vacancies, impurities, and small clusters.
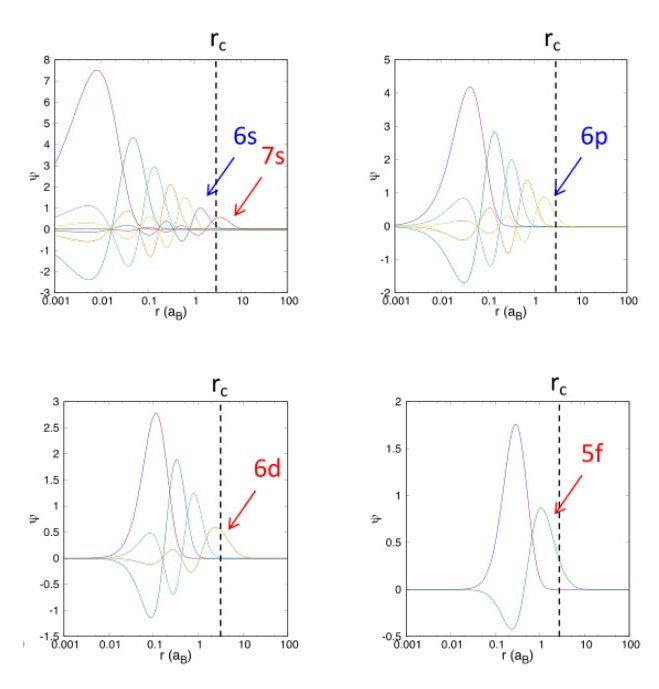
Projector augmented wave dataset: radial part of the wave function for overlapping closed shells 6s 6p and open shells 5f and 6d.
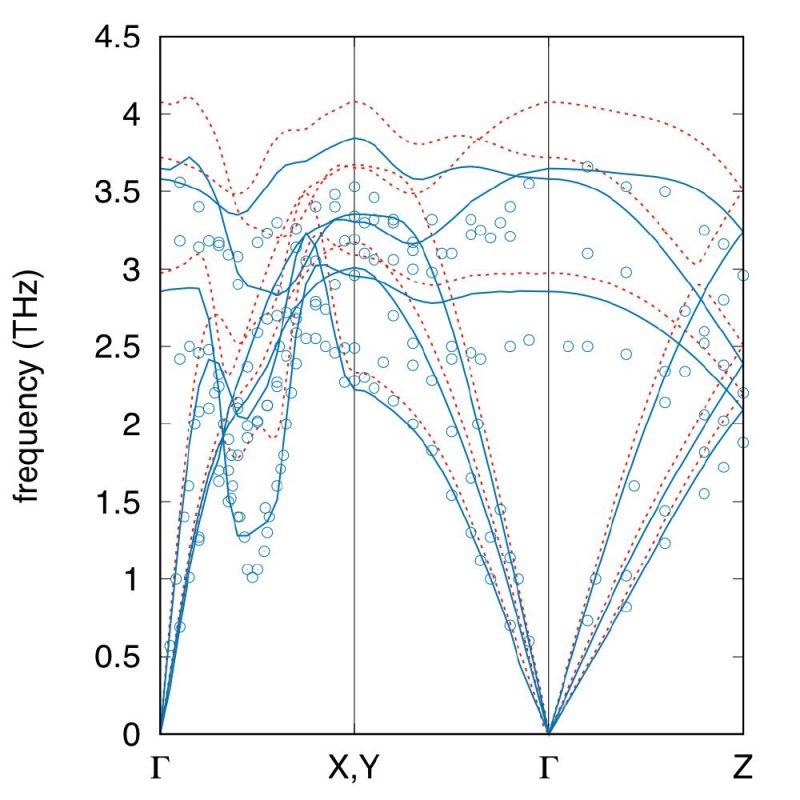
Experimental (points) and calculated (lines) phonon frequencies in α-U with Ueff=3.45eV, for atomic volumes of 20.58 (Vexp, solid blue line), and 19.89 (VLDAU, dashed red line) Å3/atom. The experimental atomic volume is 20.58 Å3/atom.
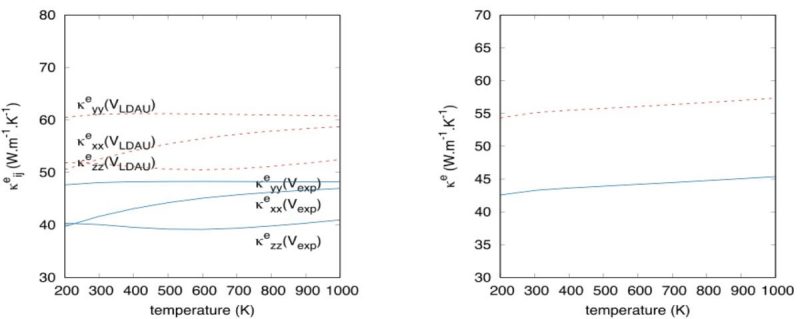
(a) Conductivity tensors in three directions. (b) Trace of conductivity tensors giving the thermal conductivity. The e-ph scattering time has been determined using ANADDB.